Edited by Nimesha Tadepalle, Ph.D., Project Coordinator at PhenoVista
Neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Huntington's disease, are characterized by the progressive loss of neurons in the brain. The underlying causes of these conditions are complex and multifactorial, with many contributing factors involved. One emerging area of research focuses on the role of lysosome dysfunction in the pathogenesis of these diseases. Here, we discuss how lysosome dysfunction impacts neurodegenerative diseases and explore the potential implications for future treatments.
TABLE OF CONTENTS
Lysosome Dysfunction in Neurodegenerative Diseases
Consequences of Lysosome Dysfunction
LYSOSOME DYSFUNCTION IN NEURODEGENERATIVE DISEASES
Lysosomes are membrane-enclosed cellular organelles responsible for the breakdown of material taken up from outside the cell, as well as waste products generated within the cell. They contain various enzymes, including proteases, lipases, and nucleases, which dismantle and recycle unneeded or damaged cellular components - a process known as autophagy. The role of lysosomes in breaking down cellular materials is key for the clearance of cellular waste and is critical in the maintenance of cellular health and homeostasis.
Research has revealed that lysosome dysfunction plays a significant role in the development and progression of neurodegenerative diseases, making it a crucial area of study. One hallmark feature of these conditions is the accumulation of toxic, protein aggregates, which can lead to cellular damage and neuronal death. Aggregation can be caused by mutations in the protein or by significant protein overexpression, which can cause misfolding that leads to protein-protein interactions. In healthy cells, lysosomes clear these aggregates through autophagy. In neurodegenerative diseases, however, lysosomal function is impaired, hindering the efficient clearance of these toxic proteins and allowing their accumulation to occur.
There are several factors that can contribute to lysosome dysfunction in neurodegenerative diseases. Genetic mutations affecting lysosomal enzymes or transporters can impair the breakdown and clearance of protein aggregates. Additionally, the activation of lysosomal enzymes can be impaired by reduced lysosomal acidification. Altered lysosomal membrane permeability can also affect the ability of lysosomes to properly degrade and clear proteins. Furthermore, impaired autophagy processes, which involve the formation and fusion of autophagosomes with lysosomes, can contribute to the accumulation of protein aggregates. Whatever the cause, the loss of these lysosomal mechanisms for breakdown and clearance of aggregated proteins ultimately contributes to the development and progression of neurodegenerative diseases.
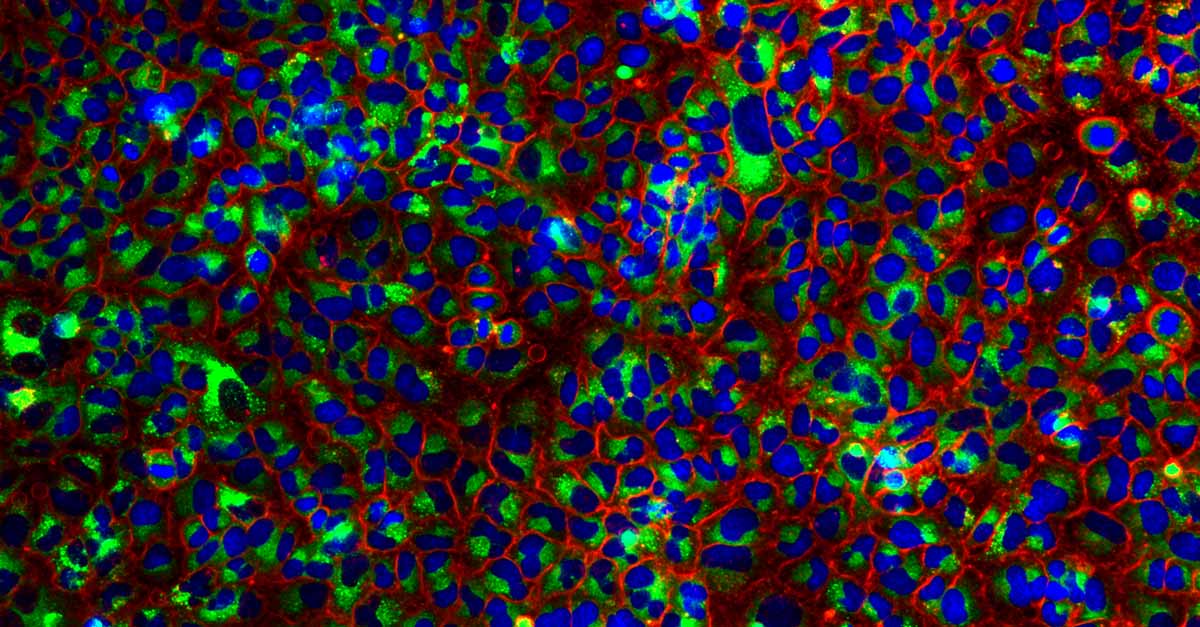
A549 cells stained for nuclei (blue), autophagosomes (red), and autolysosomes (green).
CONSEQUENCES OF LYSOSOME DYSFUNCTION
Although there are proteins common to multiple diseases, the specific protein aggregates that accumulate in neurodegenerative diseases generally vary depending on the condition. For example, in Alzheimer’s disease, the accumulation of amyloid-beta protein aggregates in the brain is a key pathological feature. Similarly, Parkinson’s disease is characterized by the accumulation of alpha-synuclein protein aggregates, while mutant huntingtin protein aggregates are associated with Huntington's disease. These aggregates not only disrupt intracellular signaling pathways, impairing neuronal communication, but they also have a detrimental impact on cognitive function, leading to cognitive decline and the manifestation of various neurological symptoms. Furthermore, these protein aggregates interfere with the proper trafficking of essential proteins within cells, compromising cellular health and contributing to neuronal dysfunction and degeneration.
Although the exact mechanisms of abnormal protein folding and subsequent aggregation are still not clearly understood, it is commonly hypothesized that oxidative stress is involved, as oxidative modification can prevent the proper protein folding. Protein aggregates can inhibit proteasome activities and stimulate reactive oxygen species (ROS) formation, resulting in oxidative damage to cellular components and further contributing to cell death. Oxidative damage exacerbates neuronal dysfunction and accelerates the loss of neurons over time.
Moreover, lysosome dysfunction and protein aggregation impair the function of mitochondria, the cellular organelles responsible for energy production. This impairment in mitochondrial function results in decreased energy production, promoting neuronal dysfunction in neurodegenerative diseases.
Another consequence of lysosome dysfunction is the triggering of chronic inflammation in neurodegenerative diseases. This chronic inflammation damages neurons and exacerbates the progression of the disease. While the immune response is intended to clear protein aggregates and protect the brain, the prolonged presence of pro-inflammatory molecules disrupts the delicate balance of neurotransmitters, impairs synaptic function, and induces oxidative stress. The oxidative stress, in turn, leads to further neuronal damage and death.
This combination of detrimental effects ultimately contributes to neurodegenerative disease presentation and progression. To address these impacts, there is a critical need for a better understanding of lysosomal dysfunction in neurodegenerative diseases and new strategies to modulate lysosomal activity. By targeting lysosome dysfunction, researchers hope to slow or halt the progression of neurodegenerative diseases and improve quality of life.
TREATMENT IMPLICATIONS
One area of research with great potential for treating neurodegenerative diseases lies in the enhancement of lysosomal function through the use of small molecules. Some of these small molecules specifically target lysosomal acidification and enzyme activity, restoring function to affected lysosomes. Similarly, small molecules that target key components of the autophagy pathway enhance the formation of autophagosomes and promote their fusion with lysosomes, promoting protein aggregate clearance through increased autophagy. By targeting multiple aspects of lysosomal dysfunction, these molecules can facilitate the clearance of protein aggregates with a multifaceted approach.
Another exciting approach is the use of gene therapy to correct genetic mutations that affect lysosomal function. Many neurodegenerative diseases are caused by mutations in genes that encode lysosomal enzymes or transporters, resulting in impaired lysosomal function and the accumulation of protein aggregates. Gene therapy aims to introduce functional copies of these genes into affected cells, either through direct delivery or the use of viral vectors. By correcting the underlying genetic defect, gene therapy aims to restore lysosomal function from its source.
The use of small molecules to improve lysosomal function and promote autophagy as well as gene therapy strategies to correct underlying mutations present promising avenues for the treatment of neurodegenerative diseases, although further research is needed to fully understand the effectiveness and potential side effects of these approaches. The advent of technologies like CRISPR/Cas9 gene editing and high-throughput screening provides powerful tools for identifying and testing potential therapeutic targets for lysosomal dysfunction. Ongoing studies in the field of neurodegenerative disease research will continue to shed light on the best therapeutic interventions for lysosome dysfunction.
CONCLUSION
Understanding the consequences of lysosome dysfunction in neurodegenerative diseases is crucial for developing effective therapies. The presence of protein aggregates not only disrupts intracellular signaling pathways and impairs cognitive function, but also triggers the generation of ROS, leading to oxidative damage and mitochondrial dysfunction. Moreover, lysosome dysfunction induces chronic inflammation, further damaging neurons and exacerbating disease progression. Currently, researchers are actively exploring various treatment strategies to enhance or restore lysosomal function and promote protein clearance.
Small molecules that target lysosomal acidification and enzyme activity, modulation of autophagy pathways, and gene therapy approaches are promising avenues for intervention. While further research is needed to fully understand their effectiveness and potential side effects, these strategies offer hope for improved quality of life for individuals with neurodegenerative diseases. By specifically targeting lysosome dysfunction and promoting protein clearance, researchers aim to restore cellular health and slow down or halt the progression of these debilitating conditions.
REFERENCES:
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat Med, 19(8), 983-997.
- Menzies FM, Fleming A, Rubinsztein DC (2015). Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci, 16(6), 345-357.
- Ballabio A, Bonifacino JS (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol, 21(2), 101-118.
- Chen X, Guo C, Kong J. (2012). Oxidative stress in neurodegenerative diseases. Neural Regen Res, 7(5), 376-85.
